Podcast
Questions and Answers
What is the primary mechanism of Tay-Sachs disease?
What is the primary mechanism of Tay-Sachs disease?
- Abnormal storage of non-metabolized products
- Defective fusion of autophagosome with lysosome (correct)
- Inability of cells to recycle damaged organelles
- Mutated enzyme responsible for breaking down fatty acids
At what age does the symptoms of Tay-Sachs disease typically begin?
At what age does the symptoms of Tay-Sachs disease typically begin?
- 3-6 months
- 1-2 years
- 3-6 years (correct)
- 6-12 months
What is the frequency of carriers among Ashkenazi Jews?
What is the frequency of carriers among Ashkenazi Jews?
- 1 in 30 (correct)
- 1 in 50
- 1 in 10
- 1 in 20
What is the typical lifespan of an individual with Tay-Sachs disease?
What is the typical lifespan of an individual with Tay-Sachs disease?
What is the characteristic of stored products in Tay-Sachs disease?
What is the characteristic of stored products in Tay-Sachs disease?
What is the underlying cause of neurologic impairment in Tay-Sachs disease?
What is the underlying cause of neurologic impairment in Tay-Sachs disease?
What is the typical order of symptoms in Tay-Sachs disease?
What is the typical order of symptoms in Tay-Sachs disease?
How does Tay-Sachs disease compare to other lipidoses?
How does Tay-Sachs disease compare to other lipidoses?
What is the primary characteristic of Gaucher cells?
What is the primary characteristic of Gaucher cells?
What is the result of the accumulation of sphingomyelin in neurons?
What is the result of the accumulation of sphingomyelin in neurons?
Which of the following organs is most severely affected in Gaucher disease?
Which of the following organs is most severely affected in Gaucher disease?
What is the term used to describe the characteristic appearance of Gaucher cells under electron microscopy?
What is the term used to describe the characteristic appearance of Gaucher cells under electron microscopy?
What is the primary lipoid that accumulates in Gaucher cells?
What is the primary lipoid that accumulates in Gaucher cells?
Which of the following is NOT a common site of involvement in Gaucher disease?
Which of the following is NOT a common site of involvement in Gaucher disease?
What is the term used to describe the enlarged neurons in Gaucher disease?
What is the term used to describe the enlarged neurons in Gaucher disease?
What is the primary function of phagocytic cells in Gaucher disease?
What is the primary function of phagocytic cells in Gaucher disease?
What is the primary characteristic of glycogenoses?
What is the primary characteristic of glycogenoses?
How many glycogenoses have been described approximately?
How many glycogenoses have been described approximately?
What is the result of complex disorders?
What is the result of complex disorders?
What is the function of the liver in glycogenoses?
What is the function of the liver in glycogenoses?
What percentage of risk is contributed by certain HLA alleles in type 1 diabetes?
What percentage of risk is contributed by certain HLA alleles in type 1 diabetes?
What is the group classification of glycogenoses based on?
What is the group classification of glycogenoses based on?
What is the primary effect of glycogenoses on glucose metabolism?
What is the primary effect of glycogenoses on glucose metabolism?
What is the approximate number of polymorphisms that may have a basis on pathophysiological findings?
What is the approximate number of polymorphisms that may have a basis on pathophysiological findings?
What is a symptom of pancreatic insufficiency related to protein absorption?
What is a symptom of pancreatic insufficiency related to protein absorption?
Which vitamins are deficient in pancreatic insufficiency?
Which vitamins are deficient in pancreatic insufficiency?
What condition may result from the inadequate synthesis of certain nutrients in infants with pancreatic insufficiency?
What condition may result from the inadequate synthesis of certain nutrients in infants with pancreatic insufficiency?
What is the result of tyrosine deficiency in children with pancreatic insufficiency?
What is the result of tyrosine deficiency in children with pancreatic insufficiency?
What is a common digestive complication of pancreatic insufficiency?
What is a common digestive complication of pancreatic insufficiency?
In which of the following is pancreatic insufficiency NOT typically associated?
In which of the following is pancreatic insufficiency NOT typically associated?
Which of the following is a potential risk as a result of protein malabsorption in infants?
Which of the following is a potential risk as a result of protein malabsorption in infants?
What physiological phenomenon can be observed due to malabsorption of fat and protein?
What physiological phenomenon can be observed due to malabsorption of fat and protein?
What primarily accumulates in Tay-Sachs disease affecting the CNS?
What primarily accumulates in Tay-Sachs disease affecting the CNS?
In Tay-Sachs disease, which cell type is primarily damaged by the accumulation of gangliosides?
In Tay-Sachs disease, which cell type is primarily damaged by the accumulation of gangliosides?
The molecular basis for neuronal injury in Tay-Sachs disease is understood to be:
The molecular basis for neuronal injury in Tay-Sachs disease is understood to be:
What protein may induce the unfolded protein response in Tay-Sachs disease?
What protein may induce the unfolded protein response in Tay-Sachs disease?
What is a key morphological characteristic of cells affected by Tay-Sachs disease?
What is a key morphological characteristic of cells affected by Tay-Sachs disease?
What type of cell death is associated with Tay-Sachs disease due to the accumulation of gangliosides?
What type of cell death is associated with Tay-Sachs disease due to the accumulation of gangliosides?
Which process may be triggered in response to the presence of misfolded proteins in Tay-Sachs disease?
Which process may be triggered in response to the presence of misfolded proteins in Tay-Sachs disease?
Which of the following best describes the affected tissues in Tay-Sachs disease?
Which of the following best describes the affected tissues in Tay-Sachs disease?
Phenylketonuria is caused by mutations that enhance the enzyme phenylalanine hydroxylase.
Phenylketonuria is caused by mutations that enhance the enzyme phenylalanine hydroxylase.
Mucous plugging and dilation of the tracheobronchial tree are not apparent in individuals with phenylketonuria.
Mucous plugging and dilation of the tracheobronchial tree are not apparent in individuals with phenylketonuria.
Phenylketonuria affects 1 in 100,000 live-born Caucasian individuals.
Phenylketonuria affects 1 in 100,000 live-born Caucasian individuals.
Phenylketonuria is more common in Jewish populations than in Scandinavian populations.
Phenylketonuria is more common in Jewish populations than in Scandinavian populations.
Phenylketonuria may be related to the involvement of one organ or many.
Phenylketonuria may be related to the involvement of one organ or many.
Pseudomonas infection is not associated with phenylketonuria.
Pseudomonas infection is not associated with phenylketonuria.
The pulmonary parenchyma is not consolidated by a combination of secretions and pneumonia in phenylketonuria.
The pulmonary parenchyma is not consolidated by a combination of secretions and pneumonia in phenylketonuria.
Phenylketonuria is an acquired error of metabolism.
Phenylketonuria is an acquired error of metabolism.
The electron microscope shows prominent lysosomes with whorled configurations in a normal neuron.
The electron microscope shows prominent lysosomes with whorled configurations in a normal neuron.
Gaucher disease is characterized by the accumulation of glucocerebroside in neurons.
Gaucher disease is characterized by the accumulation of glucocerebroside in neurons.
Niemann-Pick disease type A is characterized by early-onset CNS disease and late-onset organomegaly.
Niemann-Pick disease type A is characterized by early-onset CNS disease and late-onset organomegaly.
MPS Type I (Hurler syndrome) is characterized by the accumulation of glycogen in neurons.
MPS Type I (Hurler syndrome) is characterized by the accumulation of glycogen in neurons.
Glycogenosis type 2 (Pompe disease) is characterized by the accumulation of sphingomyelin in neurons.
Glycogenosis type 2 (Pompe disease) is characterized by the accumulation of sphingomyelin in neurons.
Lysosomal storage disorders are typically inherited in an autosomal recessive pattern.
Lysosomal storage disorders are typically inherited in an autosomal recessive pattern.
The electron microscope is not useful in diagnosing lysosomal storage disorders.
The electron microscope is not useful in diagnosing lysosomal storage disorders.
Tay-Sachs disease is characterized by the accumulation of GM1 ganglioside in neurons.
Tay-Sachs disease is characterized by the accumulation of GM1 ganglioside in neurons.
Niemann-Pick disease type C is characterized by the accumulation of cholesterol and gangliosides in neurons.
Niemann-Pick disease type C is characterized by the accumulation of cholesterol and gangliosides in neurons.
Lysosomal storage disorders are typically treatable with enzyme replacement therapy.
Lysosomal storage disorders are typically treatable with enzyme replacement therapy.
Gaucher disease is caused by a gain of function mutation in glucocerebrosidase.
Gaucher disease is caused by a gain of function mutation in glucocerebrosidase.
Mucopolysaccharides are synthesized by connective tissue cells.
Mucopolysaccharides are synthesized by connective tissue cells.
Glycogen storage diseases are caused by defects in enzyme function that break down glycogen to glucose.
Glycogen storage diseases are caused by defects in enzyme function that break down glycogen to glucose.
Gaucher disease is an X-linked dominant disorder.
Gaucher disease is an X-linked dominant disorder.
The primary characteristic of Gaucher cells is the accumulation of glycogen.
The primary characteristic of Gaucher cells is the accumulation of glycogen.
Environmental factors play a minor role in the expression of genetic disorders.
Environmental factors play a minor role in the expression of genetic disorders.
Type 2 diabetes is an example of a glycogen storage disease.
Type 2 diabetes is an example of a glycogen storage disease.
Phagocytic cells play a minor role in the degradation of glucocerebrosides.
Phagocytic cells play a minor role in the degradation of glucocerebrosides.
Glycogen storage diseases are always inherited in an autosomal dominant pattern.
Glycogen storage diseases are always inherited in an autosomal dominant pattern.
The lysosomes of phagocytic cells are responsible for the degradation of mucopolysaccharides.
The lysosomes of phagocytic cells are responsible for the degradation of mucopolysaccharides.
Gaucher disease is characterized by the accumulation of ceramide in neurons.
Gaucher disease is characterized by the accumulation of ceramide in neurons.
Enzymes are not required for glycogen breakdown.
Enzymes are not required for glycogen breakdown.
Complex disorders are always caused by a single genetic mutation.
Complex disorders are always caused by a single genetic mutation.
Glucocerebrosidase is responsible for breaking down ceramide.
Glucocerebrosidase is responsible for breaking down ceramide.
In cystic fibrosis, the CFTR protein is not properly regulated, leading to the accumulation of mucus in the airways.
In cystic fibrosis, the CFTR protein is not properly regulated, leading to the accumulation of mucus in the airways.
The ENaC channel is responsible for regulating the transport of sodium ions in the sweat ducts, playing a critical role in the symptoms of cystic fibrosis.
The ENaC channel is responsible for regulating the transport of sodium ions in the sweat ducts, playing a critical role in the symptoms of cystic fibrosis.
The CFTR protein is only found in the airways, which is why cystic fibrosis primarily affects the respiratory system.
The CFTR protein is only found in the airways, which is why cystic fibrosis primarily affects the respiratory system.
The accumulation of mucus in the airways of cystic fibrosis patients is due to the overproduction of mucus, not the inability to clear it.
The accumulation of mucus in the airways of cystic fibrosis patients is due to the overproduction of mucus, not the inability to clear it.
The water content of the mucus in the airways of cystic fibrosis patients is higher compared to that of healthy individuals.
The water content of the mucus in the airways of cystic fibrosis patients is higher compared to that of healthy individuals.
The mutations in the CFTR gene can vary in severity, leading to a spectrum of symptoms in cystic fibrosis patients.
The mutations in the CFTR gene can vary in severity, leading to a spectrum of symptoms in cystic fibrosis patients.
The CFTR protein is responsible for regulating the transport of chloride ions, but not sodium ions.
The CFTR protein is responsible for regulating the transport of chloride ions, but not sodium ions.
The accumulation of mucus in the airways is the only symptom of cystic fibrosis.
The accumulation of mucus in the airways is the only symptom of cystic fibrosis.
What structural abnormalities are observed in neurons through electron microscopy in the context of certain CNS diseases?
What structural abnormalities are observed in neurons through electron microscopy in the context of certain CNS diseases?
In which parts of the nervous system are the changes observed due to swollen ganglion cells?
In which parts of the nervous system are the changes observed due to swollen ganglion cells?
What distinctive visual feature does the retina exhibit due to the changes caused by swollen ganglion cells?
What distinctive visual feature does the retina exhibit due to the changes caused by swollen ganglion cells?
What defect causes increased chloride and sodium concentration in sweat for cystic fibrosis patients?
What defect causes increased chloride and sodium concentration in sweat for cystic fibrosis patients?
What cellular component is primarily affected in the CNS and contributes to the pathology observed via electron microscopy?
What cellular component is primarily affected in the CNS and contributes to the pathology observed via electron microscopy?
How does cystic fibrosis affect sodium and water reabsorption in the airways?
How does cystic fibrosis affect sodium and water reabsorption in the airways?
What is the significance of the 'cherry red spot' seen in the retina concerning nerve cell involvement?
What is the significance of the 'cherry red spot' seen in the retina concerning nerve cell involvement?
What is the primary consequence of dehydrated mucous layers in cystic fibrosis?
What is the primary consequence of dehydrated mucous layers in cystic fibrosis?
What changes occur in the epithelial sodium channels due to cystic fibrosis?
What changes occur in the epithelial sodium channels due to cystic fibrosis?
How does the alteration of ganglion cells in the retina relate to their function in the overall nervous system?
How does the alteration of ganglion cells in the retina relate to their function in the overall nervous system?
How does an increase in intracellular sodium relate to cystic fibrosis symptoms?
How does an increase in intracellular sodium relate to cystic fibrosis symptoms?
What role do the CFTR and ENaC play in the pathophysiology of cystic fibrosis?
What role do the CFTR and ENaC play in the pathophysiology of cystic fibrosis?
Describe a consequence of the sweat duct defect in cystic fibrosis.
Describe a consequence of the sweat duct defect in cystic fibrosis.
What is a significant result of the altered ion transport in cystic fibrosis?
What is a significant result of the altered ion transport in cystic fibrosis?
Based on the information provided, what is the primary morphological characteristic of cells affected by Tay-Sachs disease, as observed under an electron microscope?
Based on the information provided, what is the primary morphological characteristic of cells affected by Tay-Sachs disease, as observed under an electron microscope?
While lysosomal storage disorders are generally rare, the text mentions a specific category of disorders that are more common. What are these more common disorders, and what is their defining characteristic?
While lysosomal storage disorders are generally rare, the text mentions a specific category of disorders that are more common. What are these more common disorders, and what is their defining characteristic?
What is the specific accumulating metabolite in Gaucher disease?
What is the specific accumulating metabolite in Gaucher disease?
What is the primary function of the deficient enzyme or protein in Pompe disease?
What is the primary function of the deficient enzyme or protein in Pompe disease?
Explain the connection between the deficient enzyme or protein and the accumulating metabolite in MPS Type I (Hurler syndrome).
Explain the connection between the deficient enzyme or protein and the accumulating metabolite in MPS Type I (Hurler syndrome).
What is the primary cellular consequence of the accumulation of gangliosides in Tay-Sachs disease?
What is the primary cellular consequence of the accumulation of gangliosides in Tay-Sachs disease?
Given the information provided, what can be inferred about the nature of the lysosome storage disorders?
Given the information provided, what can be inferred about the nature of the lysosome storage disorders?
Based on the information provided, what is the primary clinical feature of Niemann-Pick disease, type A?
Based on the information provided, what is the primary clinical feature of Niemann-Pick disease, type A?
The text mentions "whorled configurations" in the context of lysosomes. What does this term refer to, and how does it relate to the function of lysosomes?
The text mentions "whorled configurations" in the context of lysosomes. What does this term refer to, and how does it relate to the function of lysosomes?
Explain the difference between the clinical manifestations of MPS Type I (Hurler syndrome) and MPS Type II (Hunter syndrome).
Explain the difference between the clinical manifestations of MPS Type I (Hurler syndrome) and MPS Type II (Hunter syndrome).
Explain the pathogenesis of Niemann-Pick disease, specifically addressing the role of sphingomyelinase and the resulting accumulation of aberrant products.
Explain the pathogenesis of Niemann-Pick disease, specifically addressing the role of sphingomyelinase and the resulting accumulation of aberrant products.
Distinguish between the two major types of Niemann-Pick disease, A and B, highlighting the key differences in their severity and the underlying genetic mutations.
Distinguish between the two major types of Niemann-Pick disease, A and B, highlighting the key differences in their severity and the underlying genetic mutations.
What are the symptoms commonly associated with Gaucher disease?
What are the symptoms commonly associated with Gaucher disease?
Explain the connection between the accumulation of aberrant products, such as sphingomyelin, and the secondary storage of toxic proteins and the subsequent impact on cellular function in Niemann-Pick disease.
Explain the connection between the accumulation of aberrant products, such as sphingomyelin, and the secondary storage of toxic proteins and the subsequent impact on cellular function in Niemann-Pick disease.
Identify the primary genetic cause of Gaucher disease.
Identify the primary genetic cause of Gaucher disease.
Describe the role of mitochondria in the pathogenesis of Niemann-Pick disease, highlighting their potential involvement in cellular dysfunction and the development of neurological symptoms.
Describe the role of mitochondria in the pathogenesis of Niemann-Pick disease, highlighting their potential involvement in cellular dysfunction and the development of neurological symptoms.
What role do phagocytes play in the pathology of Gaucher disease?
What role do phagocytes play in the pathology of Gaucher disease?
Explain the mechanism by which the accumulation of sphingomyelin leads to impaired breakdown of sphingomyelin into ceramide and phosphorylcholine in Niemann-Pick disease.
Explain the mechanism by which the accumulation of sphingomyelin leads to impaired breakdown of sphingomyelin into ceramide and phosphorylcholine in Niemann-Pick disease.
What type of inheritance pattern is observed in Gaucher disease?
What type of inheritance pattern is observed in Gaucher disease?
Discuss the potential therapeutic strategies that might be explored to address the underlying molecular mechanisms of Niemann-Pick disease, focusing on the role of enzyme replacement therapy and gene therapy.
Discuss the potential therapeutic strategies that might be explored to address the underlying molecular mechanisms of Niemann-Pick disease, focusing on the role of enzyme replacement therapy and gene therapy.
Which mucopolysaccharides are primarily accumulated in Gaucher disease?
Which mucopolysaccharides are primarily accumulated in Gaucher disease?
Explain the potential benefits and challenges associated with using enzyme replacement therapy to treat Niemann-Pick disease.
Explain the potential benefits and challenges associated with using enzyme replacement therapy to treat Niemann-Pick disease.
What is the primary enzymatic function of glucocerebrosidase?
What is the primary enzymatic function of glucocerebrosidase?
Describe the type of tissue typically affected by Gaucher disease.
Describe the type of tissue typically affected by Gaucher disease.
Explain the potential benefits and challenges associated with using gene therapy to treat Niemann-Pick disease.
Explain the potential benefits and challenges associated with using gene therapy to treat Niemann-Pick disease.
How does the severity of Gaucher disease manifest?
How does the severity of Gaucher disease manifest?
Azoosperma and nfertty are ound in 95% of the ______ affected
Azoosperma and nfertty are ound in 95% of the ______ affected
P umonar y d s eas e semmng rom ob s r uc on and n e c on ______ erens
P umonar y d s eas e semmng rom ob s r uc on and n e c on ______ erens
The broncoles of en are d send e d w e muc us ______ associated
The broncoles of en are d send e d w e muc us ______ associated
Clncal Features.e clncal manesaons of CF are ______
Clncal Features.e clncal manesaons of CF are ______
The primary characteristic of stored products in ______ disease is the accumulation of glucocerebroside
The primary characteristic of stored products in ______ disease is the accumulation of glucocerebroside
The ______ of the pancreas may be obstructed, leading to vitamin A deficiency.
The ______ of the pancreas may be obstructed, leading to vitamin A deficiency.
Pancreatic insufficiency may result in ______ of certain nutrients in infants
Pancreatic insufficiency may result in ______ of certain nutrients in infants
The accumulation of thick plugs of ______ can lead to pancreatic insufficiency.
The accumulation of thick plugs of ______ can lead to pancreatic insufficiency.
______ plugging and dilation of the tracheobronchial tree are not apparent in individuals with CF
______ plugging and dilation of the tracheobronchial tree are not apparent in individuals with CF
Pseudomonas ______ is associated with CF
Pseudomonas ______ is associated with CF
The ______ of pancreatic secretions can result from the dysfunction of the CFTR gene.
The ______ of pancreatic secretions can result from the dysfunction of the CFTR gene.
Meconium ileus, a type of bowel obstruction, may occur in infants due to the presence of thick ______ in the intestines.
Meconium ileus, a type of bowel obstruction, may occur in infants due to the presence of thick ______ in the intestines.
The CFTR gene regulates the transport of bicarbonate ions in the ______ cells of the exocrine pancreas.
The CFTR gene regulates the transport of bicarbonate ions in the ______ cells of the exocrine pancreas.
The ______ is an important protein involved in the regulation of chloride and bicarbonate ion transport.
The ______ is an important protein involved in the regulation of chloride and bicarbonate ion transport.
Induction of _______________ of cell death
Induction of _______________ of cell death
Pancreatic insufficiency can lead to malabsorption of fat and ______ in infants.
Pancreatic insufficiency can lead to malabsorption of fat and ______ in infants.
In Fig. 6.5, a complex ___________________ is normally degraded by a series of lysosomal enzymes
In Fig. 6.5, a complex ___________________ is normally degraded by a series of lysosomal enzymes
If there is a deficiency or malfunction of one of the enzymes, catabolism is incomplete, and insoluble ___________________ accumulate in the lysosomes
If there is a deficiency or malfunction of one of the enzymes, catabolism is incomplete, and insoluble ___________________ accumulate in the lysosomes
The presence of thick mucus plugs can lead to ______ of the pancreatic ducts.
The presence of thick mucus plugs can lead to ______ of the pancreatic ducts.
In type A, excess ____________________ accumulates in phagocytes and neurons
In type A, excess ____________________ accumulates in phagocytes and neurons
The macropahges become stuffed with ____________________
The macropahges become stuffed with ____________________
The morphology in type A is characterized by the presence of ____________________ in phagocytes and neurons
The morphology in type A is characterized by the presence of ____________________ in phagocytes and neurons
The process of cellular damage is caused by the buildup of ____________________ radicals
The process of cellular damage is caused by the buildup of ____________________ radicals
The cellular debris accumulates in the ____________________
The cellular debris accumulates in the ____________________
In cystic fibrosis (CF), a chloride channel defect in the sweat duct causes increased ______ concentration in sweat.
In cystic fibrosis (CF), a chloride channel defect in the sweat duct causes increased ______ concentration in sweat.
Patients with CF have decreased chloride secretion and increased sodium and ______ reabsorption in the airways.
Patients with CF have decreased chloride secretion and increased sodium and ______ reabsorption in the airways.
The dehydration of the mucous layer coating epithelial cells leads to defective ______ action.
The dehydration of the mucous layer coating epithelial cells leads to defective ______ action.
CFTR stands for Cystic fibrosis transmembrane ______ regulator.
CFTR stands for Cystic fibrosis transmembrane ______ regulator.
Increased intracellular sodium and abscesses are ______ in cystic fibrosis.
Increased intracellular sodium and abscesses are ______ in cystic fibrosis.
The epithelial sodium channel ENaC is responsible for intracellular sodium ______.
The epithelial sodium channel ENaC is responsible for intracellular sodium ______.
The chronic condition caused by cystic fibrosis is called chronic ______.
The chronic condition caused by cystic fibrosis is called chronic ______.
Decreased chloride secretion in CF leads to increased sodium and water ______.
Decreased chloride secretion in CF leads to increased sodium and water ______.
Some polymorphisms are associated with multiple diseases, usually liver __________ due to accumulation.
Some polymorphisms are associated with multiple diseases, usually liver __________ due to accumulation.
Glycogen forms, and __________ due to a failure of glucose production.
Glycogen forms, and __________ due to a failure of glucose production.
Many genetic variants linked to disease result from a lack of __________-6-phosphatase.
Many genetic variants linked to disease result from a lack of __________-6-phosphatase.
The __________ regions of the genome are likely associated with an hereditary form of glycogenosis.
The __________ regions of the genome are likely associated with an hereditary form of glycogenosis.
Von Gierke disease is a type I __________, a glycogen storage disease.
Von Gierke disease is a type I __________, a glycogen storage disease.
Polymorphisms may have a basis on __________ findings related to glycogen metabolism.
Polymorphisms may have a basis on __________ findings related to glycogen metabolism.
Liver diseases often demonstrate __________ enlargement when associated with glycogen accumulation.
Liver diseases often demonstrate __________ enlargement when associated with glycogen accumulation.
Glycogenoses primarily affect glucose __________ in the body.
Glycogenoses primarily affect glucose __________ in the body.
Match the following lysosomal storage diseases with their primary characteristics:
Match the following lysosomal storage diseases with their primary characteristics:
Match the following symptoms with the corresponding disease:
Match the following symptoms with the corresponding disease:
Match the following diseases with their primary effects on metabolism:
Match the following diseases with their primary effects on metabolism:
Match the following cellular features with the corresponding disease:
Match the following cellular features with the corresponding disease:
Match the following diseases with their primary organ involvement:
Match the following diseases with their primary organ involvement:
Match the following diseases with their diagnostic methods:
Match the following diseases with their diagnostic methods:
Match the following diseases with their primary consequences:
Match the following diseases with their primary consequences:
Match the following diseases with their inheritance patterns:
Match the following diseases with their inheritance patterns:
Match the following glycogenoses with their characteristics:
Match the following glycogenoses with their characteristics:
Match the following symptoms with their corresponding glycogenoses:
Match the following symptoms with their corresponding glycogenoses:
Match the following enzymes with their corresponding functions:
Match the following enzymes with their corresponding functions:
Match the following diseases with their corresponding effects on glucose metabolism:
Match the following diseases with their corresponding effects on glucose metabolism:
Match the following cell types with their corresponding characteristics:
Match the following cell types with their corresponding characteristics:
Match the following diseases with their corresponding inheritance patterns:
Match the following diseases with their corresponding inheritance patterns:
Match the following diseases with their corresponding ages of onset:
Match the following diseases with their corresponding ages of onset:
Match the following diseases with their corresponding primary organs affected:
Match the following diseases with their corresponding primary organs affected:
Match the following genetic diseases with their associated characteristics:
Match the following genetic diseases with their associated characteristics:
Match the following clinical manifestations with their respective genetic diseases:
Match the following clinical manifestations with their respective genetic diseases:
Match the following symptoms to the corresponding genetic abnormality:
Match the following symptoms to the corresponding genetic abnormality:
Match the following descriptions with the correct disease:
Match the following descriptions with the correct disease:
Match the following mechanisms with the related genetic diseases:
Match the following mechanisms with the related genetic diseases:
Match the following characteristics with their respective diseases:
Match the following characteristics with their respective diseases:
Match the following treatments with related genetic diseases:
Match the following treatments with related genetic diseases:
Match the following causal factors to their genetic disease:
Match the following causal factors to their genetic disease:
Match the metabolic disorders with their primary characteristics:
Match the metabolic disorders with their primary characteristics:
Match the following syndromes with their distinctive features:
Match the following syndromes with their distinctive features:
Match the following types of disorders with their mechanisms:
Match the following types of disorders with their mechanisms:
Match the following enzymes with their associated disorders:
Match the following enzymes with their associated disorders:
Match the following symptoms with their corresponding conditions:
Match the following symptoms with their corresponding conditions:
Match the following definitions to the terms:
Match the following definitions to the terms:
Match the following clinical features with their associated syndromes:
Match the following clinical features with their associated syndromes:
Match the following characteristics with the respective diseases:
Match the following characteristics with the respective diseases:
Match the following disorders with their primary accumulation product.
Match the following disorders with their primary accumulation product.
Match the following disorders with their primary affected organ/tissue.
Match the following disorders with their primary affected organ/tissue.
Match the following disorders with the defective enzyme.
Match the following disorders with the defective enzyme.
Match the following disorders with their characteristic cell type.
Match the following disorders with their characteristic cell type.
Match the following disorders with their mode of inheritance.
Match the following disorders with their mode of inheritance.
Match the following disorders with their typical clinical presentation.
Match the following disorders with their typical clinical presentation.
Match the following disorders with their prevalence.
Match the following disorders with their prevalence.
Match the following disorders with their treatment options.
Match the following disorders with their treatment options.
Flashcards are hidden until you start studying
Study Notes
Pancreatic Insufficiency
- Associated with malabsorption of proteins and fats, leading to large amounts of undigested intermediates excreted in urine.
- Can induce strong odors in sweat, resembling musky or "mousy" scents.
- Deficiency in fat-soluble vitamins (A, D, K) may result in hypoproteinemia and pale coloration in infants due to poor synthesis.
- Severe malabsorption can lead to generalized edema and chronic diarrhea.
Tyrosine Metabolism
- Tyrosine is essential for melanization; its deficiency can lead to neurological issues, including rectal prolapse in approximately 10% of children with cystic fibrosis.
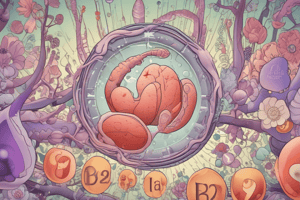
Tay-Sachs Disease
- Characterized by the accumulation of glycolipids (gangliosides) in various tissues, predominantly neurons and glial cells in the CNS.
- Molecular basis of neuronal injury is not fully understood, but misfolded proteins can trigger the unfolded protein response, leading to cell death.
- Affected cells appear swollen, sometimes foamy, due to both storage of nonmetabolized products and secondary neuronal damage.
- Normal appearance at birth; motor weakness typically starts at 3-6 months, followed by progressive neurological decline and death usually by age 2-3.
- Common among Ashkenazi Jews, with carrier frequency estimated at 1 in 30.
Gaucher Disease
- Defective fusion of autophagosomes with lysosomes leads to storage of glucocerebrosides in macrophages (Gaucher cells).
- Gaucher cells exhibit a unique "wrinkled tissue paper" appearance due to engorgement with nonmetabolized products.
- Splenomegaly is noticeable, and other affected organs include the liver, bone marrow, lymph nodes, and lungs.
- Neurons accumulate sphingomyelin, leading to further neuronal enlargement and vacuolization.
Glycogenoses
- Classified as autosomal recessive diseases, with about a dozen types described.
- Each type can be categorized into three groups based on pathophysiological findings, highlighting complex traits.
- Hepatic type glycogenoses involve defects in liver glycogen synthesis and breakdown, linking to type 1 diabetes and the impact of certain HLA alleles.
- More than 50% of the risk for specific glycogenoses may stem from genetic polymorphisms.
Cystic Fibrosis
- Characterized by abnormal chloride (Cl-) and sodium (Na+) transport in sweat ducts.
- In normal conditions, CFTR and ENaC work together to maintain electrolyte balance.
- In cystic fibrosis, mutations in CFTR lead to dehydrated mucus in the airway, causing obstruction and infection.
Phenylketonuria (PKU)
- Caused by mutations affecting the enzyme phenylalanine hydroxylase.
- Results in the accumulation of phenylalanine, leading to severe neurological impairment if untreated.
- Affects approximately 1 in 10,000 live-born Caucasian infants, more common in those of Scandinavian descent.
Lysosomal Storage Disorders
- Diseases caused by deficiencies in specific enzymes, leading to the accumulation of unmetabolized substances in cells.
- Selected disorders include:
- Tay-Sachs Disease: Deficiency in Hexosaminidase causing GM2 ganglioside accumulation; primarily affects the CNS.
- Gaucher Disease: Deficiency in Glucocerebrosidase leading to glucocerebroside accumulation; manifests as organomegaly and CNS disease.
- Niemann-Pick Disease: Caused by sphingomyelinase deficiency, resulting in sphingomyelin accumulation; presents early-onset CNS disease in Type A and organomegaly in Type B.
- MPS Type I (Hurler Syndrome) and Type II (Hunter Syndrome): Deficiency in lysosomal enzymes leads to the accumulation of heparin and dermatan sulfate; associated with organomegaly and cardiovascular disease.
- Pompe Disease (Glycogenosis Type 2): Due to deficiency in acid maltase; characterized by glycogen accumulation causing cardiac failure.
Pathogenesis of Disorders
- Lysosomal storage disorders often result from defective enzymes critical for metabolizing specific substances.
- Accumulation leads to various clinical manifestations including organomegaly, CNS symptoms, and motor regressions.
- Environmental factors can influence the expressivity of genetic disorders, contributing to variability in symptom severity.
General Considerations
- Most lysosomal storage disorders are rare, with some more prevalent in specific populations due to genetic background.
- Continuous research is essential to understand the detailed mechanisms and potential therapies for these genetic diseases.
Cystic Fibrosis (CF)
- CF is characterized by a defect in chloride channels, specifically affecting the CFTR protein.
- Increased chloride and sodium concentrations in sweat are diagnostic indicators of CF.
- In the airways, CF leads to decreased chloride secretion and heightened sodium and water reabsorption.
- Dehydration of the mucous layer results in impaired mucociliary action and mucous plugging.
- Increased intracellular sodium absorption from dysfunctional ENaC channels contributes to chronic bronchitis and bronchiectasis.
- Common lung complications include abscess formation due to thickened mucus and blockage of airways.
- Electron microscopy shows abnormal whorled membrane configurations and lysosomal changes throughout the central and peripheral nervous system.
- The retina is affected, with swollen ganglion cells causing a "cherry-red" spot on the unaffected central macula.
Niemann-Pick Disease
- Caused by mutations affecting acid sphingomyelinase, leading to sphingomyelin accumulation.
- Type A is more severe, primarily affecting the central nervous system, while Type B presents with organomegaly and later-onset CNS symptoms.
- Clinical features include significant developmental delays and physical abnormalities.
Lysosomal Storage Disorders
- Tay-Sachs disease involves a deficiency in hexosaminidase, causing GM2 ganglioside accumulation, primarily impacting the CNS.
- Gaucher disease is caused by a deficiency in glucocerebrosidase, leading to variable presentations: mild forms cause organomegaly, while severe forms lead to CNS disease.
- Niemann-Pick disease has variants with differing symptoms; Type C involves NPC1 or NPC2 deficiencies, presenting with neurological symptoms and variable organomegaly.
- Mucopolysaccharidoses (MPS) involve enzyme deficiencies (e.g., Hurler and Hunter syndromes), leading to heparin and dermatan sulfate accumulation with varying severity.
- Pompe disease results from a deficiency in acid maltase, leading to glycogen accumulation and cardiac issues.
Pathogenesis Overview
- Lysosomal storage disorders are rare but cause significant cellular dysfunction due to enzyme deficiencies.
- Accumulation of toxic metabolites leads to cellular degeneration and prompts secondary inflammatory responses, observable under microscopy as prominent lysosomal structures.
Cystic Fibrosis Overview
- Cystic fibrosis (CF) results from a defect in the chloride channel found in sweat ducts.
- Elevated levels of chloride and sodium are observed in the sweat of CF patients.
- In airways, CF leads to reduced chloride secretion alongside increased sodium and water reabsorption.
- Dehydration of the mucous layer damages epithelial cells, impairs mucociliary action, and causes mucus plugging.
Impact of CF on Respiratory System
- CFTR (Cystic fibrosis transmembrane conductance regulator) and ENaC (epithelial sodium channel) are critical in sodium transport regulation.
- Increased sodium absorption contributes to chronic bronchitis and bronchiectasis.
- Lung development is severely affected, leading to respiratory complications.
Pancreatic Complications
- Thickened secretions may obstruct pancreatic ducts, resulting in pancreatic insufficiency.
- Vitamin A deficiency may arise from blocked ducts and reduced transport of bicarbonate ions in the exocrine pancreas.
- Meconium ileus, a type of bowel obstruction, may occur due to thick plugs in intestines.
Male Fertility Issues
- Azoospermia and infertility occur in 95% of affected males, primarily due to the absence of the vas deferens.
- Males with CF may still survive to adulthood despite the reproductive impact.
Clinical Manifestations
- Clinical features of CF include respiratory symptoms due to mucus build-up and bacterial infections.
- Bronchioles often show dilatation due to obstructive mucus and associated hypertrophy.
Pathology of Mucus Production
- Excess mucus production leads to cell damage and inflammation.
- Morphological changes in macrophages and neurons occur due to cellular stress and damage from free radicals.
Genetic and Disease Relationships
- Some polymorphisms may link to multisystemic disorders, notably liver enlargement from the accumulation of substrates.
- Abnormal glycogen metabolism can lead to specific diseases, including hypoglycemia due to failure in glucose production.
Enzyme Deficiencies and Disease Mechanism
- Deficiencies in lysosomal enzymes can lead to incomplete catabolism of substrates, resulting in the accumulation of harmful metabolites within lysosomes.
- Specific gene variants associated with noncoding regulatory regions of the genome may contribute to the pathogenesis of certain diseases, including CF.
Liver Disease in Pregnant Women
- Significant risk of liver disease arises in the later stages of pregnancy.
- Management necessitates a low phenylalanine diet prior to conception due to teratogenic implications.
- Liver disease has become the third most prevalent cause of mortality in expectant mothers, overshadowed by pulmonary and pancreatic conditions.
Lysosomal Storage Diseases
- Result from mutations impacting lysosomal enzyme functionality, leading to metabolic degradation issues.
- Diagnosis often suspected with persistent elevations in enzyme deficiencies.
- Characteristic clinical findings include pulmonary complications and gastrointestinal manifestations.
- Cystic Fibrosis (CF) patients may face additional complications related to organ transplantation.
Tay-Sachs Disease
- Most common gangliosidosis, attributed to mutations affecting hexosaminidase A enzyme expression.
- Causes lysosomal enzyme deficiency, resulting in impaired degradation pathways.
- Clinical presentation typically includes neurodegenerative symptoms associated with glycolipid accumulation.
Gaucher Disease
- Involves variable bone-related symptoms such as osteopenia, osteoporosis, corneal clouding, and mental disability.
- Characterized by lipid-laden Gaucher cells in bone marrow and other tissues, which can be observed through microscopy.
- Identifiable by the presence of distinct clinical features and may lead to significant morbidity.
Mucopolysaccharidoses (MPS)
- Several clinical variants exist, often recognized by the accumulation of glycosaminoglycans due to enzyme deficiencies.
- Hurler syndrome (MPS type I) caused by α-L-iduronidase deficiency leads to severe systemic complications, reducing life expectancy to 6 to 10 years.
- Hunter syndrome (MPS type II) differs with a later onset, more gradual progression, and X-linked inheritance.
- Both syndromes exhibit corneal clouding and cardiovascular implications, but in Hunter syndrome, enzyme replacement can mitigate cardiac issues.
Glycogen Storage Diseases
- Associated with muscle weakness due to glycogen accumulation in muscle tissue.
- McArdle disease (Type V) causing elevated blood lactate levels is commonly found in athletes.
- Enhanced likelihood of cardiac complications and myoglobinuria following physical exertion due to muscle injury.
Complex Multigenic Disorders
- Arise from the interplay of multiple genetic variants and environmental triggers.
- The genesis of complex disorders showcases how various genes collaboratively contribute to disease manifestations.
Studying That Suits You
Use AI to generate personalized quizzes and flashcards to suit your learning preferences.